SÍNDROME DE ALPORT: UN TRASTORNO GENÉTICO CON IMPACTO MULTISISTÉMICO
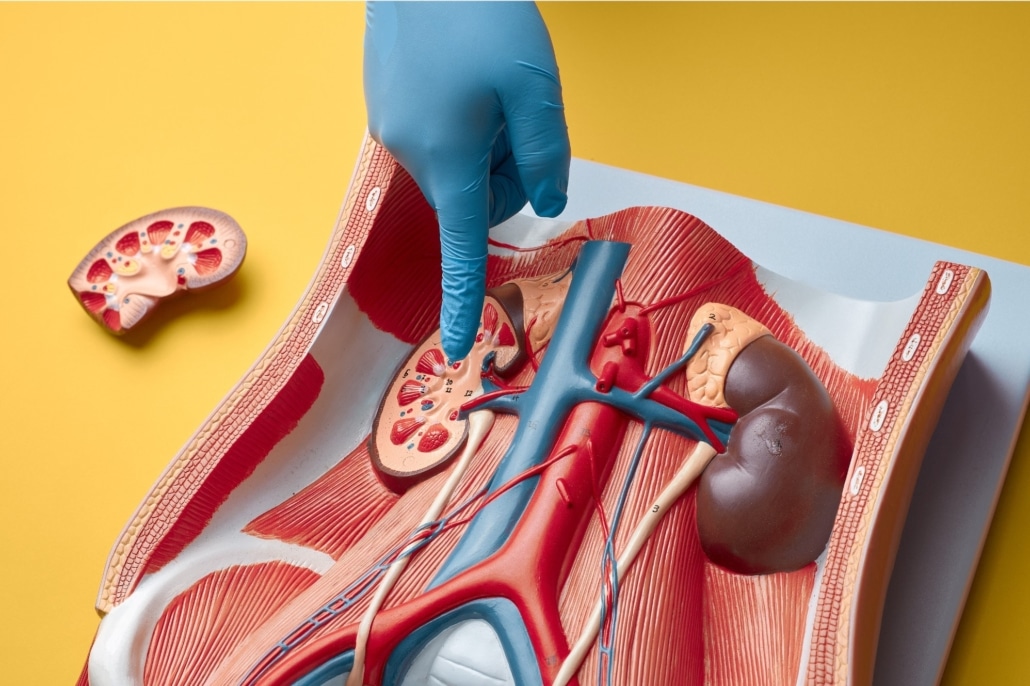
El síndrome de Alport es una enfermedad genética que afecta principalmente a los riñones, los oídos y los ojos. Se caracteriza por una progresiva disfunción renal, pérdida de audición neurosensorial y, en algunos casos, alteraciones oculares. Este trastorno, aunque relativamente raro, tiene un impacto significativo en la calidad de vida de los pacientes y sus familias. En este artículo, exploraremos en profundidad las causas, síntomas, diagnóstico y opciones de tratamiento del síndrome de Alport.
Origen genético del Síndrome de Alport
El síndrome de Alport es causado por mutaciones en los genes que codifican las cadenas de colágeno tipo IV, una proteína esencial para la estructura y función de las membranas basales en los riñones, los oídos y los ojos. Este colágeno forma una red especializada que brinda soporte estructural y regula el transporte de moléculas. Las mutaciones afectan la integridad de esta red, debilitando las membranas basales y causando los síntomas característicos de la enfermedad.
Los genes implicados incluyen:
-COL4A5: Este gen se localiza en el cromosoma X y es el principal responsable del síndrome de Alport ligado al X, que representa la mayoría de los casos. Las mutaciones en COL4A5 suelen resultar en una disrupción significativa del colágeno tipo IV, especialmente en los hombres, quienes portan solo una copia del cromosoma X.
-COL4A3 y COL4A4: Estos genes están localizados en el cromosoma 2 y están relacionados con las formas autosómicas recesiva y dominante de la enfermedad. Las mutaciones en COL4A3 o COL4A4 pueden causar síntomas menos severos en algunos casos y en las forma recesivas normalmente ambos padres son portadores silentes de una copia defectuosa del gen.
La variedad de mutaciones observadas en estos genes incluye deleciones, inserciones, sustituciones y errores de empalme, que afectan la producción y ensamblaje del colágeno. Esto conduce a una fragilidad estructural en las membranas basales, lo que resulta en la disfunción de los órganos afectados.
Además, las diferencias en la expresión clínica entre los pacientes están relacionadas con el tipo de mutación y la forma de herencia. Por ejemplo, las mutaciones truncantes en COL4A5 suelen asociarse con una progresión rápida hacia insuficiencia renal terminal, mientras que las mutaciones menos severas pueden permitir un curso más lento de la enfermedad.
La comprensión de estas bases genéticas no solo es clave para el diagnóstico preciso, sino también para el desarrollo de terapias personalizadas que apunten a corregir o mitigar los efectos de estas mutaciones.
Manifestaciones clínicas
El síndrome de Alport afecta múltiples sistemas en el cuerpo, y sus manifestaciones clínicas pueden variar ampliamente dependiendo de la forma genética y la severidad de la enfermedad.
Afectación renal
La afectación renal es el signo más prominente del síndrome de Alport. Los pacientes suelen presentar:
-Hematuria persistente: Es uno de los primeros signos y puede detectarse desde la infancia.
-Proteinuria: Su presencia indica un deterioro progresivo de la función renal.
-Insuficiencia renal crónica: Los pacientes avanzan hacia insuficiencia renal terminal (ERT), especialmente los hombres con la forma ligada al X.
En algunos casos, el inicio temprano de la hematuria y la velocidad de progresión hacia la ERT pueden ser utilizados como marcadores pronósticos para determinar la severidad de la enfermedad.
Pérdida auditiva
La pérdida de audición neurosensorial afecta aproximadamente al 80% de los pacientes antes de los 30 años. Generalmente comienza afectando las frecuencias altas, lo que puede dificultar la comprensión del habla, especialmente en ambientes ruidosos. Este síntoma tiene un impacto significativo en la calidad de vida, ya que interfiere con la comunicación diaria y el desarrollo social.
Alteraciones oculares
Aunque menos comunes, las alteraciones oculares pueden ser diagnósticas en algunos pacientes:
-Lenticono anterior: Una deformación del cristalino que puede llevar a problemas de refracción y pérdida de agudeza visual.
-Retinopatía: Incluye manchas o puntos pigmentados en la retina.
-Adelgazamiento de la membrana basal de la retina: Detectable mediante exámenes oftalmológicos especializados.
Estas manifestaciones pueden ser detectadas mediante estudios de imagen ocular y son útiles como parte del diagnóstico diferencial.
Otros síntomas
En casos raros, el síndrome de Alport puede estar asociado con:
-Aneurismas intracraneales: Reportados en un pequeño subgrupo de pacientes.
-Dolores musculares o articulares: Probablemente relacionados con la fragilidad de las membranas basales en otros tejidos.
Diagnóstico
El diagnóstico del síndrome de Alport se realiza combinando herramientas clínicas, genéticas y de laboratorio. Una evaluación integral incluye:
1.Historia clínica detallada: Incluyendo antecedentes familiares de enfermedad renal o pérdida auditiva.
2.Análisis de orina: Para identificar hematuria y proteinuria.
3.Biopsia renal: Proporciona información clave sobre la estructura de las membranas basales.
4.Pruebas genéticas: Identifican mutaciones en COL4A3, COL4A4 o COL4A5, confirmando el diagnóstico.
5.Exámenes audiológicos y oftalmológicos: Para evaluar las manifestaciones en oídos y ojos.
El diagnóstico temprano permite implementar intervenciones que retrasen la progresión de la enfermedad y mejoren los resultados a largo plazo.
Tratamiento y manejo
No existe una cura para el síndrome de Alport, pero las estrategias de tratamiento están orientadas a manejar los síntomas y prevenir complicaciones:
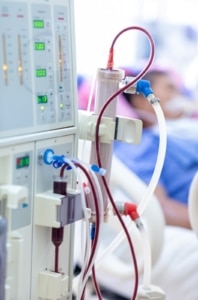
Tratamiento renal
-Inhibidores de la enzima convertidora de angiotensina (IECA): Reducen la proteinuria y protegen la función renal.
-Diálisis y trasplante renal: Opciones para pacientes con ERT. Los resultados del trasplante suelen ser favorables, aunque en algunos casos pueden desarrollarse anticuerpos anti-colágeno tipo IV.
Manejo auditivo
–Uso de audífonos para mejorar la audición en etapas tempranas.
-Implantes cocleares: En casos de pérdida auditiva severa.
Manejo Ocular
–Monitorización regular por un oftalmólogo para detectar alteraciones como el lenticono.
–Corrección refractiva si es necesario.
Es esencial para educar a las familias sobre el riesgo de transmisión y las opciones reproductivas, como el diagnóstico genético preimplantacional.
Conclusión
El síndrome de Alport es un trastorno genético multisistémico que impacta principalmente los riñones, los oídos y los ojos, comprometiendo significativamente la calidad de vida de quienes lo padecen. Si bien no existe una cura, el diagnóstico temprano y el manejo integral pueden retrasar la progresión de la enfermedad y mejorar los resultados a largo plazo.
La identificación temprana de signos como hematuria persistente, pérdida auditiva progresiva y alteraciones oculares, junto con pruebas genéticas, es esencial para un tratamiento eficaz. Intervenciones como el uso de inhibidores de la ECA, dispositivos auditivos y el asesoramiento genético ofrecen herramientas valiosas para abordar los síntomas y prevenir complicaciones.
Con un enfoque preventivo y personalizado, es posible mitigar los efectos del síndrome de Alport y brindar a los pacientes una mejor calidad de vida, enfatizando la importancia del seguimiento médico regular y el apoyo familiar.
Laboratorio Lorgen Genética y Proteómica.