NEUROFIBROMATOSIS: UNA PERSPECTIVA DERMATOLÓGICA
La neurofibromatosis es un grupo de trastornos genéticos caracterizados por la predisposición a formar tumores en el sistema nervioso. Se divide en tres tipos principales: neurofibromatosis tipo 1 (NF1), neurofibromatosis tipo 2 (NF2) y schwannomatosis. De estos, la NF1 es la más común y tiene una amplia gama de manifestaciones clínicas, especialmente dermatológicas, que pueden ser indicativas del trastorno y esenciales para su diagnóstico temprano.
Neurofibromatosis Tipo 1 (NF1)
La NF1, también conocida como enfermedad de Von Recklinghausen, afecta aproximadamente a 1 de cada 3,000 personas. Esta enfermedad autosómica dominante se origina por mutaciones en el gen NF1, localizado en el cromosoma 17, que codifica la neurofibromina, una proteína que actúa como supresora de tumores. Aproximadamente la mitad de los casos de NF1 son heredados, mientras que la otra mitad resulta de mutaciones de novo.
Manifestaciones dermatológicas de NF1
Las manifestaciones dermatológicas son uno de los aspectos más visibles y diagnósticos de la NF1. Entre las principales se encuentran:
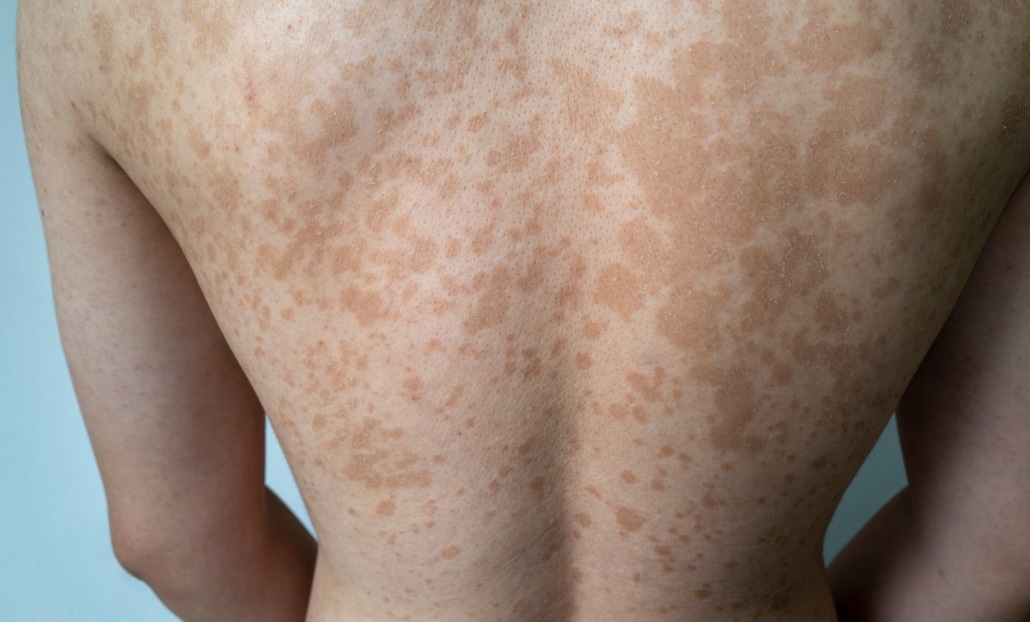
MANCHAS CAFÉ CON LECHE (CAFÉ-AU-LAIT)
Estas manchas son lesiones hiperpigmentadas planas que varían en tamaño desde unos pocos milímetros hasta varios centímetros. Su color varía de marrón claro a oscuro, similar al café con leche, de ahí su nombre. La presencia de seis o más manchas café con leche mayores de 5 mm en preadolescentes, o mayores de 15 mm en adolescentes y adultos, es un criterio diagnóstico clave para NF1.
NEUROFIBROMAS
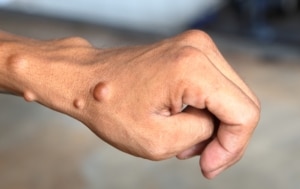
-Neurofibromas dérmicos: Son tumores benignos que se desarrollan en la piel. Pueden ser solitarios o múltiples y varían en tamaño. A menudo, los neurofibromas dérmicos son blandos y de color piel o ligeramente más oscuros. Aunque generalmente no son dolorosos, pueden ser desfigurantes.
-Neurofibromas subcutáneos: Se encuentran debajo de la piel y pueden causar molestias o dolor si comprimen estructuras nerviosas cercanas.
-Neurofibromas plexiformes: Estos son tumores más grandes y complejos que pueden afectar nervios más grandes y tejidos circundantes. Son congénitos y pueden causar deformidades significativas y problemas funcionales. Además, tienen un riesgo aumentado de transformación maligna en tumores de la vaina del nervio periférico maligno (MPNST, por sus siglas en inglés).
EFÉLIDES AXILARES E INGUINALES
Estas son pequeñas manchas pigmentadas que aparecen en áreas de pliegues cutáneos como las axilas y la ingle. Son un signo distintivo y ayudan en el diagnóstico de NF1.
NÓDULOS DE LISCH
Son hamartomas benignos del iris, visibles mediante examen con lámpara de hendidura. No afectan la visión, pero son un hallazgo ocular característico de NF1 y pueden ayudar en el diagnóstico.
GLIOMAS ÓPTICOS
Aunque estos tumores no son manifestaciones cutáneas, son comunes en NF1 y pueden causar pérdida de visión si no se detectan y tratan a tiempo. Su presencia es relevante para una evaluación completa de la enfermedad.
Diagnóstico y evaluación
El diagnóstico de NF1 se basa principalmente en criterios clínicos bien definidos. Además de las manifestaciones dermatológicas, otros hallazgos clínicos y familiares son importantes para confirmar el diagnóstico.
CRITERIOS DIAGNÓSTICOS DE NF1
Para diagnosticar NF1, se requiere la presencia de dos o más de los siguientes criterios:
Seis o más manchas café con leche de al menos 5 mm de diámetro en preadolescentes, o 15 mm en adolescentes y adultos.
-Dos o más neurofibromas de cualquier tipo o un neurofibroma plexiforme.
-Pecas en la región axilar o inguinal.
-Glioma del nervio óptico.
-Dos o más nódulos de Lisch.
-Una lesión ósea característica, como displasia del esfenoides o pseudoartrosis.
-Un pariente de primer grado con NF1 diagnosticado según estos criterios.
Tratamiento y manejo
El manejo de la NF1 es complejo y requiere un enfoque multidisciplinario debido a la variedad de sistemas que puede afectar. Los dermatólogos, junto con neurólogos, oncólogos, oftalmólogos y otros especialistas, juegan un papel crucial en el diagnóstico y manejo de esta condición.
OPCIONES DE TRATAMIENTO
Observación y monitoreo: Debido a la naturaleza progresiva de NF1, es esencial un seguimiento regular para monitorear la aparición de nuevos tumores y la progresión de los existentes. Los exámenes anuales que incluyan evaluaciones dermatológicas, neurológicas y oftalmológicas son recomendables.
Intervención quirúrgica: Los neurofibromas que causan dolor, son desfigurantes o presentan un riesgo de transformación maligna pueden ser removidos quirúrgicamente. Sin embargo, la cirugía puede ser complicada por la localización y la recurrencia es común.
Tratamientos farmacológicos: Aunque no hay tratamientos curativos específicos aprobados para NF1, se están investigando diversas terapias dirigidas. Los inhibidores de mTOR (rapamicina) y MEK han mostrado promesas en estudios preclínicos y ensayos clínicos tempranos.
Terapias de apoyo: Los pacientes pueden beneficiarse de terapias físicas y ocupacionales para manejar las complicaciones musculoesqueléticas. Además, el apoyo psicológico es vital debido al impacto emocional y social de la enfermedad.
Investigación y avances
La comprensión de las bases moleculares de la neurofibromatosis tipo 1 (NF1) ha avanzado significativamente en las últimas décadas, especialmente a partir de la identificación del gen NF1 y su producto proteico, la neurofibromina. Este descubrimiento ha sido fundamental para entender la patogénesis de la enfermedad y para el desarrollo de nuevas estrategias terapéuticas.
El gen NF1 está ubicado en el cromosoma 17q11.2 y codifica una proteína conocida como neurofibromina, que actúa como un supresor tumoral. La neurofibromina desempeña un papel crucial en la regulación de la vía de señalización Ras, una cascada molecular que controla varios procesos celulares, incluidos el crecimiento y la proliferación celular. En condiciones normales, la neurofibromina inactiva la proteína Ras, previniendo así la proliferación celular descontrolada.
Sin embargo, las mutaciones en el gen NF1 resultan en una pérdida de función de la neurofibromina. Esto conduce a la activación constante de la vía Ras, lo que provoca un crecimiento celular excesivo y la formación de tumores. Esta hiperactivación de la vía Ras es un mecanismo central en la patogénesis de NF1, y ha sido un foco de investigación para desarrollar terapias dirigidas.
Conclusión
La neurofibromatosis tipo 1 es una enfermedad compleja con diversas manifestaciones dermatológicas que pueden tener un impacto significativo en la calidad de vida de los pacientes. Un enfoque multidisciplinario que incluya dermatólogos, genetistas, neurólogos y otros especialistas es crucial para el manejo efectivo de esta condición. La investigación en curso ofrece esperanza para el desarrollo de terapias más efectivas en el futuro, subrayando la importancia de continuar los esfuerzos científicos y clínicos en esta área. Con una mayor comprensión de la enfermedad y avances en la medicina personalizada, el pronóstico para los pacientes con NF1 puede mejorar significativamente en los próximos años.
Laboratorio Lorgen Genética y Proteómica.