
HEMOFILIA: UN TRASTORNO HEREDITARIO QUE AFECTA LA COAGULACIÓN
La hemofilia es un trastorno hemorrágico hereditario que afecta al proceso de coagulación sanguínea. Se debe a la falta o mal funcionamiento de proteínas específicas: el factor VIII en la hemofilia A y el factor IX en la hemofilia B. Estas proteínas plasmáticas son esenciales para una coagulación adecuada. Cuando están ausentes o no funcionan correctamente, se produce una reducción y un retraso en la formación de trombina, una proteína clave en la coagulación. Como resultado, se generan defectos en la formación de coágulos, lo que provoca episodios de sangrado que pueden afectar tejidos blandos, articulaciones y músculos.
La clasificación de la gravedad de la hemofilia depende de la cantidad de factores de coagulación VIII o IX que están activos y funcionando correctamente:

Prevalencia
La hemofilia afecta a la población por igual, independientemente de su origen étnico o geográfico. Se estima que tiene una frecuencia de uno de cada 5000 a 10000 nacidos vivos. La hemofilia A afecta aproximadamente a 1 de cada 5000 nacimientos masculinos a nivel mundial (80% de los casos de hemofilia), mientras que la hemofilia B afecta a alrededor de 1 de cada 30000 nacimientos masculinos.
Genética y bases moleculares
La hemofilia A y B son enfermedades causadas por mutaciones (cambios en la secuencia de ADN) en los genes que producen los factores de coagulación VIII y IX, respectivamente. Estas mutaciones heredadas afectan la producción o la función de estos factores de coagulación, provocando los síntomas observados en los pacientes con hemofilia.
En la hemofilia A, las mutaciones en el gen F8 son responsables de la producción defectuosa o incompleta del factor VIII. En casos graves de hemofilia A, las mutaciones más comunes suelen causar un codón de parada prematuro o afectar al proceso de «splicing» (edición del gen). Alrededor del 45% de las mutaciones graves se encuentran en una región del gen conocida como intrón 22.
El factor VIII viaja en la sangre unido al factor von Willebrand (FvW), que lo protege de ser degradado. En el proceso de coagulación, el factor VIII activa el factor X y mejora la capacidad del factor IX para producir trombina, esencial para la formación de coágulos.
Por otro lado, la hemofilia B se debe a mutaciones en el gen F9, siendo la mayoría pequeños cambios en la secuencia del ADN. Las mutaciones que generan un codón de parada son frecuentes en casos graves.
El factor IX, al igual que el factor VIII, es crucial en la coagulación sanguínea. Es una enzima que, al activarse, colabora con el factor X para producir trombina.
Herencia
Tanto la hemofilia A como la hemofilia B son trastornos de herencia recesiva ligada al cromosoma X. Esto significa que los genes responsables de estos trastornos se encuentran en el cromosoma X. En las mujeres, que tienen dos cromosomas X (XX), normalmente ambas copias del gen deben estar alteradas para causar la enfermedad. Si una mujer hereda una copia normal y una mutada, será portadora del trastorno y generalmente no mostrará síntomas, debido a la presencia de la copia normal del gen en el otro cromosoma X. En cambio, en los hombres, que solo tienen un cromosoma X (XY), una única copia mutada del gen es suficiente para desarrollar la enfermedad.
Aun así, en las mujeres, en algunos casos una sola copia alterada del gen F8 o F9 puede ser suficiente para desarrollar la enfermedad, porque el cromosoma X con la copia normal del gen se desactiva, en un proceso denominado inactivación del cromosoma X. Este proceso ocurre temprano en el desarrollo y asegura que tanto mujeres como hombres tengan activo solo un cromosoma X en las células de su organismo. Por lo general, este proceso es aleatorio (cada cromosoma X está activo en la mitad de las células), pero a veces un cromosoma X está activo en más de la mitad de las células (inactivación sesgada).
Para las mujeres con una alteración en una copia del gen F8 o F9 y una inactivación aleatoria, alrededor de la mitad de las células tendrán activo el cromosoma X con la copia normal del gen, lo que suele ser suficiente para producir niveles adecuados de factor de coagulación. En cambio, en mujeres con inactivación sesgada, el cromosoma X con la copia normal del gen puede estar inactivado en más de la mitad de las células. Esto puede resultar en niveles insuficientes de factor de coagulación VIII o IX, aumentando el riesgo de hemorragias.
Debido a este tipo de herencia, la hemofilia afecta casi exclusivamente a hombres. Las mujeres normalmente son portadoras y pueden mostrar síntomas leves debido a niveles reducidos de factor VIII o IX.
Diagnóstico

El diagnóstico de la hemofilia es de gran importancia para un adecuado tratamiento y la identificación de las mujeres portadoras. En general, los pacientes con hemofilia grave son diagnosticados antes de los 2 años, mientras que aquellos con hemofilia leve pueden pasar varios años sin ser diagnosticados, ya que algunos solo presentan síntomas cuando tienen lesiones o se someten a cirugías.
Para diagnosticar la hemofilia, se realizan pruebas para medir la actividad de los factores de coagulación FVIII y FIX. Estas pruebas incluyen el ensayo de una etapa, que compara el tiempo de coagulación del paciente con el de un plasma de referencia, y el ensayo cromogénico, que mide la cantidad de FX activado para evaluar la función de los factores FVIII o FIX.
El análisis genético para identificar la mutación responsable de la enfermedad se realiza principalmente en casos graves y moderados de hemofilia. En formas leves, donde los síntomas son menores, no suele ser necesario por su costo y porque el diagnóstico prenatal no siempre es crucial.
En el caso de la hemofilia B, se realiza secuenciación del gen completo F9. Para la hemofilia A, el primer paso es buscar inversiones en los intrones 22 e 1 del gen F8, responsables de aproximadamente el 40% y el 1% de los casos severos, respectivamente, seguido de la secuenciación completa del gen.
Tratamiento
El tratamiento principal para la hemofilia implica la administración del factor de coagulación que falta para mantener niveles adecuados y prevenir o detener los sangrados. Hay dos enfoques principales en la terapia: la profilaxis, que previene los sangrados antes de que ocurran, y la terapia a demanda, que trata el sangrado cuando se presenta. Además de estos enfoques tradicionales, se están investigando y estudiando nuevas estrategias:
-Factores de coagulación de semivida extendida: estos factores, al tener mayor duración en el organismo, permiten reducir la frecuencia de administración y hemorragias, con respecto al tratamiento tradicional.
-Agentes no sustitutivos que mejoran la coagulación: un ejemplo es el Emicizumab, un anticuerpo que facilita la coagulación al unir las proteínas de coagulación factor IX activado y factor X.
-Tratamiento no sustitutivo; inhibiendo los inhibidores: un método que utiliza RNA de interferencia para bloquear la producción de una proteína llamada antitrombina en el hígado. Esto ayuda a que el cuerpo produzca más trombina, lo que mejora la coagulación.
-Terapia génica: este enfoque busca curar la hemofilia introduciendo genes que producen los factores de coagulación directamente en el hígado. Se utilizan virus modificados para llevar estos genes a las células del hígado, donde pueden empezar a producir estos factores de manera constante.
Hemofilia adquirida
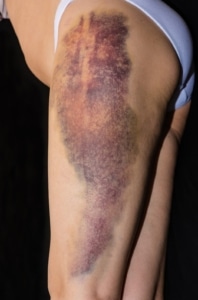
La hemofilia adquirida es una enfermedad rara de origen autoinmune que afecta aproximadamente a entre 1 y 1,5 personas por cada millón al año. Esta cifra aumenta a 15 casos por millón en personas mayores de 85 años. En esta condición, el sistema inmunológico produce anticuerpos que atacan los factores de coagulación naturales, causando episodios de sangrado espontáneo, que suelen ser graves. Aunque es más frecuente en hombres mayores de 85 años, también puede manifestarse en mujeres jóvenes con enfermedades autoinmunes, durante el embarazo o después del parto.
En resumen, la hemofilia es un trastorno hemorrágico hereditario causado por la deficiencia o mal funcionamiento de los factores de coagulación VIII (hemofilia A) o IX (hemofilia B). Estos factores son fundamentales para la formación de coágulos sanguíneos, y su ausencia o disfunción lleva a episodios de sangrado que afectan tejidos blandos, articulaciones y músculos. La hemofilia se hereda de manera recesiva ligada al cromosoma X y afecta principalmente a hombres. El diagnóstico precoz y el tratamiento con factores de coagulación son cruciales para manejar los síntomas y prevenir complicaciones graves.
Patricia Sánchez Fariña. Graduada en Bioquímica por la Universidad de Sevilla y estudiante del Máster en Biología Molecular Aplicada a Empresas Biotecnológicas.
Bibliografía