DIAGNÓSTICO GENÉTICO PRENATAL INVASIVO: CASO CLÍNICO
El diagnóstico prenatal permite orientar a la familia sobre el pronóstico, planificar un adecuado seguimiento obstétrico, ofrecer un manejo intrauterino para casos seleccionados poco comunes, y derivar a la mujer embarazada a un centro de atención terciaria que pueda proporcionar diagnóstico y tratamiento neonatal. Esto ha demostrado reducir la morbilidad y mortalidad perinatales asociadas (Hautala et al., 2019; Meller et al., 2020).
En la práctica clínica actual nos encontramos con varias opciones de pruebas genéticas prenatales disponibles para mujeres embarazadas. No obstante, este servicio sólo ha estado disponible en los últimos años a pesar de que el afán por la detección prenatal de anomalías cromosómicas lleva en curso desde la década de 1960. Al principio, la edad materna era prácticamente el único parámetro de cribado elegido y la trisomía 21 la anomalía cromosómica que se examinaba casi exclusivamente (Grande et al., 2012). Las pruebas genéticas prenatales en fetos han experimentado una larga evolución desde los métodos invasivos tradicionales como son la amniocentesis, el análisis de vellosidades coriónicas y la cordocentesis a los menos invasivos como es el análisis del ADN fetal libre en sangre materna (Palomaki et al., 2011; Bringman, 2014).
Caso clínico: exploración ecográfica, antecedentes y motivo del estudio

Para poder entender cómo llevamos a cabo un estudio genético prenatal empleando varias técnicas, se presenta el caso de una paciente embarazada a la cual el especialista detectó en el feto por ecografía: una malformación cardíaca de canal auriculoventricular (AV) completo junto con hipertrofia renal bilateral, aumento de ecogenicidad y un tamaño fetal menor del esperado para su edad (feto PEG). Además, la paciente presentaba antecedentes familiares previos ya que anteriormente dio a luz mediante cesárea a un recién nacido vivo que presentaba hipoplasia del ventrículo izquierdo, más conocido como síndrome del corazón izquierdo hipoplásico (SCIH). Finalmente, el recién nacido falleció a las pocas horas de vida.
Al tratarse de un caso de alto riesgo se decidió someter a la paciente, tras informarla y dar su consentimiento, a una amniocentesis ecoguiada transplacentaria obteniendo muestra de líquido amniótico suficiente para poder realizar así el estudio genético en el feto. Además, también se obtuve muestra de sangre periférica materna para comparar y descartar cualquier contaminación materna en la muestra de líquido amniótico.
Pruebas genéticas realizadas y resultados obtenidos
Identifiler
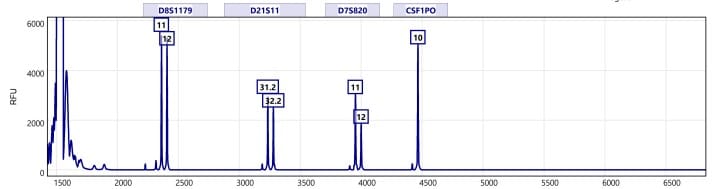
Se realizó un Identifiler para descartar alguna posible contaminación de células maternas en la muestra de líquido amniótico mediante la comparación de 16 marcadores genéticos de tipo STR del kit AmpFlSTR Identifiler Plus (Applied Biosystems) con la muestra de sangre materna. El análisis realizado mostró un perfil genético único y de sexo femenino (XX) en la muestra de líquido amniótico.
Array prenatal
Se llevó a cabo un array prenatal para analizar las ganancias y pérdidas de material genético (copy number variation (CNV)) utilizando el chip CytoScan OPTIMA prenatal (ThermoFisher®).
Tras realizar el análisis de la dotación genética de la muestra de líquido amniótico y salvando las limitaciones técnicas que se indicaron, se informó de lo siguiente:
-Fórmula genómica según nomenclatura ISCN (2020): arr(X,1-22)x2. Lo que equivale a 2 cromosomas sexuales X y 44 autosomas completos.
-La muestra analizada presentaba un patrón genómico de sexo femenino (XX).
-No se detectaron alteraciones de tipo CNV de naturaleza no polimórfica.
Exoma clínico dirigido
Por otro lado, se secuenció el exoma completo para así poder analizar las mutaciones puntuales. Concretamente usando los datos brutos del exoma completo secuenciado se realizó a través del análisis bioinformático un exoma clínico dirigido a cardiopatías y anomalías renales congénitas. Se tuvieron en cuenta los genes incluidos en un panel customizado de Lorgen GP, en los paneles de PanelApp «miocardiopatía pediátrica o sindrómica (Versión 3.12)» y «miocardiopatía hipertrófica (Versión 4.4)», así como en los siguientes fenotipos: cardiomiopatía (HP:0001638), cardiomiopatía hipertrófica (HP:0001639) hipoplasia renal (HP:0000089) y displasia renal (HP:0000110).
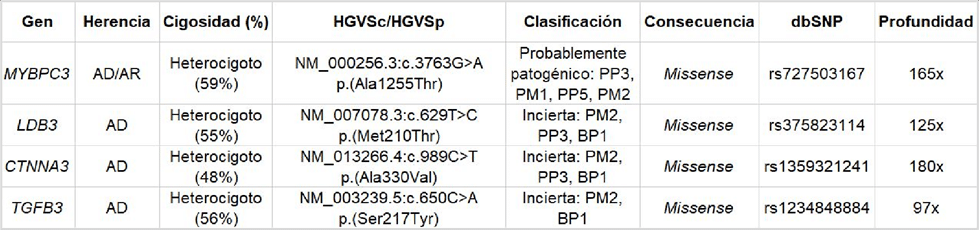
Se detectó en heterocigosis la variante relacionada con cardiopatías congénita y de significado clínico probablemente patogénico, según los criterios de la American College of Medical Genetics (ACMG): c.3763G>A p.(Ala1255Thr) en el gen MYBPC3. Además, se encontraron otras variantes de significado clínico incierto (VUS), según los criterios de la ACMG.
Se detectó en homocigosis la variante relacionada con anomalías renales congénitas y de significado clínico patogénico, según los criterios de la ACMG: c.118del p.(Cys40AlafsTer2) en el gen BBS1.

Alteraciones en el gen MYBPC3 se relacionan con cardiomiopatía dilatada 1MM y no compactación ventricular izquierda 10 (OMIM:615396), de herencia autosómica dominante. Una miocardiopatía debida a la parada de la morfogénesis del miocardio y caracterizada por un ventrículo izquierdo hipertrófico, un miocardio de 2 capas severamente engrosado, numerosas trabeculaciones prominentes profundas, recesos intertrabeculares y función sistólica deficiente (Probst et al., 2011). También con miocardiopatía hipertrófica 4 (OMIM:115197), de herencia tanto autosómica dominante como recesiva. Un trastorno cardíaco hereditario caracterizado por hipertrofia ventricular, que suele ser asimétrica y que a menudo afecta al tabique interventricular (Wang et al., 2013). Por otra parte, alteraciones en el gen BBS1 se relacionan con el síndrome de Bardet-Biedl 1 (OMIM:209900) caracterizado por malformación renal y de manera secundaria por cardiopatías congénitas (Melluso et al., 2023). Para las VUS encontradas, las patologías asociadas a los genes en los que se encuentran presentan herencia autosómica dominante. Sin embargo, la evidencia disponible actualmente es insuficiente para determinar el papel de esas VUS en la clínica del feto.
Conclusión
Podemos concluir que encontramos la posible base genética de la clínica que presentaba el feto y que los resultados obtenidos tienen implicaciones hereditarias y familiares. Estos resultados junto con otros tipos de pruebas pueden orientar al especialista en cuanto a qué medidas tomar. Se recomienda un adecuado asesoramiento genético en el contexto familiar concreto, llevar a cabo una reclasificación de las VUS informadas pasado al menos un año para comprobar si su significado clínico se ha esclarecido, así como realizar un estudio de cosegregación/segregación familiar para las variantes detectadas. Por otra parte, a pesar de ya haber encontrado posibles variantes genéticas puntuales que explicarían la clínica del feto también se podría, pasado un tiempo considerable y de ser necesario, efectuar un reanálisis en busca de nuevas variantes ya que guardamos los datos brutos generados de la ultrasecuenciación del exoma completo.
Francisco Javier Moya García, graduado en Biología. Analista Genético en Lorgen GP.
Referencias
- Bringman Invasive prenatal genetic testing: A Catholic healthcare provider’s perspective. Linacre Q 2014;81:302–313. SAGE Publications Sage CA: Los Angeles, CA.
- Hautala J, Gissler M, Ritvanen A, Tekay A, Pitkänen-Argillander O, Stefanovic V, Sarkola T, Helle E, Pihkala J, Pätilä T, et al. The implementation of a nationwide anomaly screening programme improves prenatal detection of major cardiac defects: an 11-year national population-based cohort BJOG Int J Obstet Gynaecol 2019;126:864–873.
- Meller CH, Grinenco S, Aiello H, Córdoba A, Sáenz-Tejeira MM, Marantz P, Otaño Congenital heart disease, prenatal diagnosis and management. Arch Argent Pediatr 2020;118:e149–e161.
- Melluso A, Secondulfo F, Capolongo G, Capasso G, Zacchia Bardet-Biedl Syndrome: Current Perspectives and Clinical Outlook. Ther Clin Risk Manag 2023;19:115–132.
- Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, Boom D van den, Bombard AT, Deciu C, Grody WW. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation Genet Med 2011;13:913–920. Nature Publishing Group.
- Probst S, Oechslin E, Schuler P, Greutmann M, Boyé P, Knirsch W, Berger F, Thierfelder L, Jenni R, Klaassen S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet 2011;4:367–